Desórdenes orales y carcinoma oral de células escamosas: ¿Por qué el diagnóstico, el pronóstico y la terapia asociados con el cáncer oral permanecieron sin cambios?
El carcinoma de células escamosas (CCE) es el más común de todas las neoplasias malignas orales. Se ha observado que entre el 11% y el 36% de leucoplasias con displasia y que hasta el 70% de leucoplasias verrucosas proliferativas pueden convertirse en lesión maligna cada año(1,2.)
La etiología del carcinoma oral de células escamosas (COCE) ha sido considerada multifactorial(3,4). Específicamente, se diagnostican más de 300.000 casos de cáncer oral por año alrededor del mundo, incluyendo labio, los cuales 50% no llegan con vida a los 5 años. El sur de Brasil, Argentina y Uruguay tienen las mayores tasas de incidencia de cáncer de boca y faringe en América Latina(5). Debido a su ubicación, los carcinomas orales y faríngeos también pueden estar asociados con una morbilidad significativa, ya que estos cánceres y tratamientos pueden ser mutiladores y afectar las funciones diarias, lo que impacta negativamente a la calidad de vida. Las tasas de supervivencia se han mantenido relativamente sin cambios durante las últimas tres décadas, probablemente debido al reconocimiento tardío de la enfermedad, como ocurre en la mayoría de los países(4). En consecuencia, se hace cada vez más destaque al diagnóstico precoz y monitoreo frecuente de las lesiones precancerosas.
A pesar de estos esfuerzos, los esquemas actuales de clasificación no logran diferenciar con precisión los desórdenes sin potencial progresivo de los con potencial progresivo, siendo así el manejo clínico de los pacientes con estas lesiones no debiera basarse en este criterio exclusivo. Por otro lado, varios estudios han demostrado una gran variabilidad inter e intra-examinador en la evaluación de la presencia o ausencia y el grado de la displasia epitelial oral.(6-12) Por todo lo anterior, este trabajo busca revisar qué alteraciones genéticas podrían estar involucradas en el proceso de malignización del epitelio oral, discutiendo la posible utilización de estas variaciones en el diagnóstico personalizado y terapias específicas.
Blancos terapéuticos del COCE
COCE en etapas tempranas a moderadas son frecuentemente tratados quirúrgicamente con radioterapia asociada, además, con o sin administración de quimioterapia en el contexto de adyuvante postoperatorio para pacientes de alto riesgo que tienen ganglios linfáticos comprometidos y/o metástasis que se extienden más allá de la cápsula del ganglio linfático(13-14). En la enfermedad avanzada, los enfoques multidisciplinarios no-quirúrgicos se utilizan cada vez con mayor frecuencia para mejorar el control de la enfermedad, prolongar la supervivencia y mantener una calidad de vida aceptable para los pacientes(15-17). Aunque se utilice la mejor combinación de abordajes terapéuticos, más del 50% de los pacientes con COCE experimentarán recidiva, ya sea localmente, en los ganglios linfáticos regionales o en un sitio distante(14,18). Independientemente de la ubicación o etapa del COCE, se necesitan terapias más efectivas.
La secuenciación del exoma completo de tumores primarios frente a las muestras de metástasis ha sido limitado hasta el momento debido a la falta de muestras, sin embargo, se ha observado que las lesiones metastásicas individuales son de origen clonal y son genéticamente únicas, entretanto, tienen una ascendencia clonal rastreable del tumor primario. Por lo tanto, la diversidad genética subclonal es clave para el éxito o fracaso de la terapia(19). Este es un desafío considerable técnico y bioinformático en la genómica del cáncer, que requerirá profunda secuenciación e investigación de los genomas de los patrones de segregación de mutaciones para comprender la diversidad genética dentro de neoplasias y cómo esto cambia en respuesta a las intervenciones.
Una estrategia promisora para el tratamiento del COCE y otros cánceres es la terapia dirigida molecular. En este enfoque, las alteraciones moleculares específicas de una célula cancerosa que contribuyen al fenotipo neoplásico se emplean como dianas de anticuerpos, moléculas pequeñas y/o construcciones genéticas. Este enfoque difiere de las terapias citotóxicas convencionales, que inhiben la actividad mitótica de todas las células en división, dando lugar a efectos tóxicos en todos los tejidos que normalmente tienen una rápida renovación celular, como la mucosa oral, los folículos pilosos y revestimiento intestinal(20). Se piensa que la terapia dirigida ofrece un índice terapéutico más alto y por lo tanto se asocia con menos toxicidad que los fármacos citotóxicos. El desarrollo de enfoques dirigidos al COCE requiere comprensión de la patogénesis molecular de la enfermedad, así como una mejor caracterización de los eventos moleculares específicos implicados en el crecimiento, invasión y metástasis del cáncer.
Presentación Clínica de los Desórdenes potencialmente malignos de la mucosa oral
Los desórdenes potencialmente malignos de la mucosa oral (DPMO), incluyen leucoplasia, eritroplasia, liquen plano, fibrosis submucosa oral, lupus eritematoso discoide y queilitis actínica. Las lesiones más frecuentes que preceden al desarrollo del COCE, son leucoplasia y eritroplasia(21-24). El diagnóstico clínico de estas lesiones puede ser complejo y se necesita experiencia adecuada para diferenciar clínicamente estas lesiones de otras patologías. Se han sugerido métodos clínicos complementarios(21,25-26), pero hasta el momento no se ha determinado un método no invasivo que permita identificar el potencial de riesgo de transformación maligna de una determinada DPMO.
La tasa de transformación maligna de DPMO a COCE varía según la población, sus hábitos y localización de la lesión principalmente (27). El riesgo más alto afecta a los fumadores que han utilizado cigarrillos sin filtrar por muchos años. El alcohol puede actuar como promotor, teniendo un efecto sinérgico con el tabaco(28). DPMO en piso de boca y lengua tienen un riesgo aumentado de transformación maligna, así como la edad avanzada(27).
Neoplasia sin displasia
La patología clásica enfatiza que las lesiones neoplásicas se caracterizan por anomalías arquitectónicas y citológicas que lo distinguen del tejido normal. Las características citológicas de la neoplasia incluyen agrandamiento nuclear e hipercromatismo, membranas nucleares irregulares, aumento de la proporción núcleo-citoplasma de las células, patrón de cromatina agrupada, nucléolos múltiples, elongación de núcleos, mitosis atípicas y citoplasma hipereosinofílico o denso(26,29). Muchas de estas características histopatológicas representan la expresión fenotípica de la pérdida de control de la proliferación y diferenciación celular, como resultado de mutaciones en las vías moleculares que regulan estos aspectos esenciales del crecimiento celular(19,29). Sin embargo, paralelamente a los avances recientes, la sincronización y secuencia de eventos moleculares que conducen a la transformación neoplásica, es de nuestro conocimiento de que muchas importantes anomalías moleculares que conducen a la desregulación de la proliferación y diferenciación celular ocurren antes de la expresión morfológica de la neoplasia. Por lo tanto, no todas las lesiones precursoras neoplásicas muestran necesariamente características morfológicas de displasia epitelial (Figura 1). Las alteraciones en el contenido de DNA, el control del ciclo celular y las mutaciones en los genes supresores de tumores, tales como p53 y p16, se producen en una etapa temprana de la transformación neoplásica, antes del desarrollo de características morfológicas convencionales de displasia epitelial.(29)
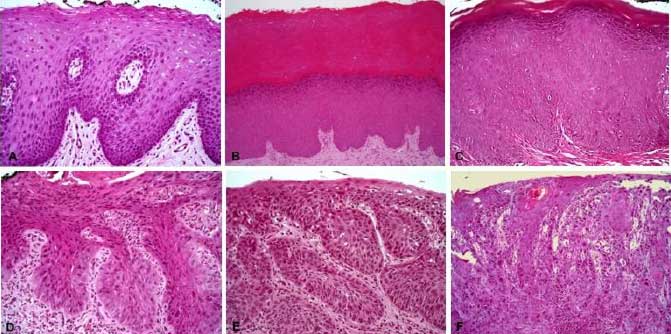
Figura 1. Cambios histológicos en la mucosa oral observados en las diferentes etapas de la transformación maligna. A. Mucosa de aspecto normal que presenta acantosis sin atipia; B. Mucosa hiperqueratinizada con acantosis e hipergranulosis; C. Mucosa queratinizada que presenta displasia epitelial de bajo grado; D. Mucosa que presenta displasia de alto grado; E. Carcinoma de células escamosas microinvasor; F. Carcinoma de células escamosas bien diferenciado que invade la lámina propia.
En muchos procesos carcinogénicos, la transformación citológica de las células representa una aberración biológica tardía en la vía carcinogénica que conduce en última instancia a la transformación de una célula normal a maligna. Por lo tanto, con mejorías en nuestra comprensión sobre alteraciones moleculares involucradas en carcinogénesis oral y con la realización de estudios que ayuden a correlacionar genotipo y fenotipo, los patólogos podrán reconocer la presencia de lesiones pre-malignas, que no necesariamente muestran evidencia morfológica de displasia epitelial.(29)
Evolución Clonal
El cáncer es una enfermedad de evolución dinámica, estocástica somática genómica(30-31). Se cree que todos los cánceres surgen como resultado de (i) inestabilidad genómica somática, (ii) generación de diversidad genómica somática, y (iii) selección natural de variantes que subyacen la progresión a cáncer(28). Basado principalmente en estudios de cánceres avanzados en un solo punto en el tiempo, se ha inferido que diferentes tipos de inestabilidad genómica tienen diferentes tasas de progresión. Por ejemplo, se cree que el cáncer colorrectal y otros cánceres se desarrollan a través de la acumulación gradual de mutaciones puntuales a una tasa de mutación normal durante décadas(32-33), lo que proporcionaría una ventana temporal prolongada de oportunidad para la detección temprana. La carcinogénesis multi-etapa es un proceso evolutivo en el que la inestabilidad genética genera nuevos clones y la selección natural entre tales variantes impulsa expansiones clonales(34).
El número de lesiones genéticas necesarias para producir un cáncer es desconocido para la mayoría de los tejidos, pero se ha estimado que es de 2 a 12(35). Las tasas normales de mutación en ausencia de expansión clonal no pueden explicar la acumulación de tantas lesiones(36-38). Tanto inestabilidad genética y expansiones clonales se observan comúnmente en la progresión neoplásica, y es probable que los dos factores colaboren en la evolución clonal(39). Si esto es cierto, entonces la expansión de un clon genéticamente inestable en una lesión premaligna se asociará con el riesgo de progresión al cáncer y, por lo tanto, es posible extraer estos marcadores en el marco del desarrollo de blancos terapéuticos.
La secuenciación del genoma del cáncer, facilitada por la introducción de la segunda generación de secuenciación del genoma completo, ha proporcionado una mejor visión de la complejidad genética y de biología evolutiva de las células cancerosas(40). En la mayoría de los casos, la transformación y metástasis son probablemente clonales, porque se derivan de células individuales(40). Por lo tanto, la identificación de las mutaciones presentes en todas las células de un tumor puede ayudar a reconstruir el genotipo de la célula precursora. Estos eventos precursores limitan la complejidad genética y clonal de los tumores. Ya tenemos una larga lista de mutaciones recurrentes en muchos tipos de tumores como resultado del mapeo fino de roturas cromosómicas, secuenciación de genes candidatos y selección funcional de muestras a granel de tumores.(41)
Transformación Maligna
La transformación de células normales en células cancerosas es un proceso evolutivo. Las poblaciones de células precancerosas se reproducen, mutan y compiten por los recursos. Algunas de estas mutaciones conducen eventualmente al cáncer. Aunque cada cáncer se deriva de una sola célula normal, la población de células neoplásicas que constituyen el cáncer final a menudo tiene una historia evolutiva compleja. Se cree que requieren múltiples ondas de expansión clonal, provocadas cada una por una mutación adicional del controlador, para generar el clon dominante que se manifiesta como el cáncer sintomático. A lo largo del camino, los clones de ramificación adicionales con otros controladores pueden haber sido separados por giro que no pudieron superar al clon dominante, el cual por sí mismo pudo haber producido otro clon menor con una mutación adicional del controlador que con el tiempo dominaría. Algunos de estos clones menores pueden haber sido completamente extinguidos, pero otros pueden persistir.(42-44)
Las mutaciones somáticas adquiridas por células cancerosas a medida que se dividen pueden servir como marcadores de origen clonal y permitir así la reconstrucción retrospectiva del árbol evolutivo de cánceres individuales. Estos análisis han revelado la compleja estructura clonal de ciertos cánceres y han demostrado que los clones menores en la presentación inicial del cáncer son a menudo fuente del clon mayor que se repite después del tratamiento(19,42-44). Gran parte del enfoque en investigación genómica del cáncer ha sido comprendido en los genes y vías genéticas de cánceres avanzados para mejorar las estrategias de tratamiento del paciente(45-46). Sin embargo, se ha dedicado relativamente poca investigación a la comprensión de la dinámica de la evolución genómica somática que conduce al cáncer y subyace a las observaciones clínicas donde algunas lesiones orales progresan rápidamente al COCE, mientras que el resto no progresa durante toda la vida. (Figura 2).
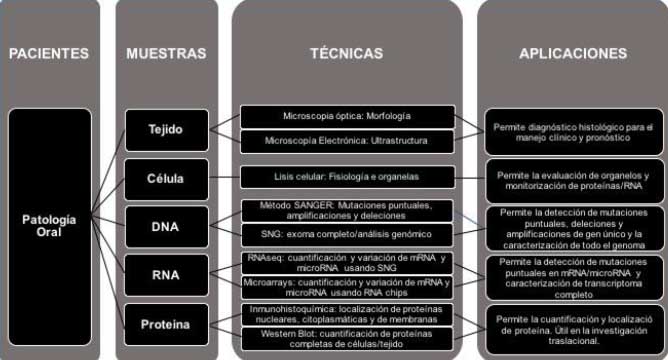
Figura 2. Presentación esquemática de la enfermedad del paciente, diagnóstico e investigación translacional a lo largo de la aplicación de técnicas moleculares en patología oral.
Muestras fijadas en formalina y embebido en parafina (FFEP)
El uso de muestras de tejido incluido, fijado en formalina e embebido en parafina (FFEP) para análisis de expresión de RNA ha recibido recientemente interés como resultado de técnicas mejoradas para recuperar ácidos nucleicos de muestras de FFEP, así como el gran número de estas muestras a menudo disponibles a los investigadores. El uso de muestras FFEP para el análisis de expresión de RNA requiere superar varios desafíos. La fijación en formalina resulta en la degradación y fragmentación del DNA/RNA, así como la modificación de los nucleótidos, todos los cuales disminuyen el rendimiento de los ácidos nucleicos extraídos de dichas muestras.(47-55)
Se ha demostrado que la mayoría de los estudios genómicos en muestras FFEP tienen resultados muy similares a las muestras frescas congeladas. Esto es de vital importancia, ya que una extracción de ácidos nucleicos de forma óptima y reproducible en estas muestras nos puede permitir ampliar el estudio genómico de enfermedades específicas cuyos resultados permitirían correlacionar aspectos clínicos, histopatológicos y ser de gran utilidad para tomar decisiones acerca de los posibles tratamientos de estas enfermedades.(56-58)
Secuenciación del exoma completo como potente herramienta para descifrar el espectro mutacional
La disección del tejido tumoral y/o pre-maligno y la secuenciación del exoma siguen siendo un área poco explorada en biología oral. La secuenciación de nueva generación (SNG) acelera el proceso de estudio del DNA mediante la generación de datos digitales y cuantificables que pueden ser asignados al genoma. La SNG puede realizar un seguimiento de las alteraciones somáticas y hereditarias responsables por la expresión de RNAm aberrantes. Considerando la heterogeneidad de las lesiones orales, SNG puede ser ideal para estudiar y comprender las alteraciones genómicas que resultan en la formación de este grupo de tumores. Los hallazgos de los estudios con SNG de lesiones orales nos ayudarían a comprender mejor los aspectos genéticos de un tumor tradicionalmente considerado ambiental. Como un análisis sinérgico, se podrá mapear de nuevo los perfiles de RNAm alterados dentro de las regiones genómicas en que se encuentran mutaciones.
Las condiciones genéticas que han sido difíciles de diagnosticar debido a variantes raras o condiciones que tienen heterogeneidad genética sustancial, han estimulado la adopción de SNG en diferentes contextos clínicos(59-60). En oncología, SNG se ha utilizado para la identificación de variantes raras y para lograr altas tasas de detección. Trujillano et al.61 evaluó recientemente el uso de un enfoque de enriquecimiento dirigido, combinado con la SNG multiplexada de secuenciación por síntesis en el análisis molecular de la fibrosis quística. Los autores informaron una tasa de detección de mutaciones de 100% y tasa de diagnóstico de 98,9%, y demostraron la capacidad de SNG para detectar grandes deleciones, duplicaciones e inversiones. Utilizando el analizador de DNA sistema MySeq se ha demostrado un rendimiento comparable, con una especificidad ≥ 99,99% en comparación con los métodos de referencia tradicionales. La alta exactitud de la SNG ofrece la promesa de detectar incluso variantes muy poco frecuentes para un gran número de enfermedades genéticas. Una vez que SNG es una clave para la obtención de objetivos diagnósticos, pronósticos y terapéuticos, estos marcadores deben ser validados en la evolución secuencial: Mutación – expresión de RNAm aberrante – cambios en niveles de proteínas(61).
Se ha demostrado en varios tipos de tumores que el estado mutacional de los pacientes es clave en la determinación de la respuesta o en los niveles de respuesta al tratamiento(62-63). El impulso de los ámbitos clínicos y de investigación para mejorar las tecnologías existentes ha dado lugar a la producción de una nueva generación de plataformas SNG con mayor precisión, eficacia en el tiempo y costo-efectivo. Se han desarrollado nuevos tipos de instrumentos de secuenciación que permiten una aceleración de las tasas de recopilación de datos para la secuenciación del DNA. La SNG ofrece la opción de producir más de mil millones de pares de bases en un trabajo de 4 días, pero se necesitan métodos sofisticados de bioinformática. El análisis y la traducción de estos datos en la clínica se complican aún más por la naturaleza heterogénea de la mayoría de los cánceres. Además, la mayoría de las muestras tumorales archivadas se conservan como tejido FFEP; Por lo tanto, la calidad del material de partida para estos estudios puede verse comprometida. A pesar de estas limitaciones, algunos estudios han utilizado con éxito muestras FFEP para estudiar mutaciones, variación del número de copias y determinación de variaciones de la línea germinal.(64-66)
Se espera que la SNG se expanda de una técnica enfocada en cáncer/mutación para incluir translocación, alteraciones del número de copias del gen y cambios epigenéticos del genoma. Con los avances en tecnología y nuestra comprensión sobre heterogeneidad intra-tumoral, la muestra inicial y el control de la línea germinal también pueden cambiar. Sin embargo, entre la producción de datos de SNG y la aplicación de estos hallazgos a las necesidades clínicas cotidianas, hay algunos obstáculos importantes a superar. En primer lugar, la detección de una alteración genética no necesariamente la convierte en una mutación iniciadora o driver. Un paso más cerca de la aplicación clínica, una mutación identificada no es necesariamente prevenible ni definitivamente tratable. Mutaciones de silenciamiento en los genes supresores de tumores son muy complejas a la inversa, y en muchos casos sus inhibidores no pueden ser usados(67). La mayoría de las vías de señalización relacionadas con cáncer son muy críticas, y la inhibición o alteración de éstas podría ser letal; sin embargo, la presencia de vías de compensación hace que la letalidad sintética sea una estrategia exitosa(68).
La heterogeneidad del carcinoma de células escamosas de cabeza y cuello, combinado con diferencias étnicas y geográficas en el genoma humano(69), requiere una atención extrema a los detalles en el diseño de estudios con SNG. SNG tiene potencial para transformar el tratamiento de atención en salud oral, prediciendo activamente el riesgo y previniéndolos. Sin embargo, la optimización y estudios adicionales detallados son necesarios para arrojar luz sobre la patogénesis molecular del cáncer para ayudar a los pacientes en situaciones clínicas cotidianas de manera personalizada y significativa.
Consideraciones Finales
Por lo tanto, la nueva generación de clínicos de salud bucal debe reconocer que el término "cáncer de cabeza y cuello" no refleja adecuadamente la complejidad de este grupo de tumores de una manera como la defendida por Ellis y Perou(69) en relación al cáncer de mama. En el cáncer de mama, desde principios del año 2000 ya se conocen por lo menos 5 grupos heterogéneos de patologías, todas definidas como cáncer de mama. Sin embargo, tanto el perfil mutacional como transcripcional difiere los subgrupos de estos tumores, La SNG es una herramienta potente, con costos cada vez más reducidos y presentes en muchos laboratorios de genética y biología molecular. El acceso de pacientes con patologías complejas incluyendo neoplasias malignas y benignas más que nunca pueden hacerse uso de esta tecnología. Por ejemplo, logramos conocer qué mutaciones iniciadoras están presentes en tejidos clínicamente benignos pero alterados y atribuir características genéticas que se asocien con el potencial de malignizarse. Como se ha señalado anteriormente, la aplicación de todas las tecnologías disponibles no hace que nuestra atención de pacientes sea personalizada, sino que nos ayuda a llegar a un "diagnóstico real" y una "etiología de concordancia para un tratamiento adecuado" que se convierte en atención estándar.(70)
Referencias
- Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015; 136(5): E359-86.
- Warnakulasuriya S, Ariyawardana A. Malignant transformation of oral leukoplakia: a systematic review of observational studies. J Oral Pathol Med. 2016; 45(3):155-66.
- Bouda M, Gorgoulis VG, Kastrinakis NG, Giannoudis A, Tsoli E, Danassi-Afentaki D et al. "High risk" HPV types are frequently detected in potentially malignant and malignant oral lesions, but not in normal oral mucosa. Mod Pathol. 2000; 13(6):644-53.
- Mashberg, A. Diagnosis of early oral and oropharyngeal squamous carcinoma: obstacles and their amelioration. Oral Oncol. 2000; 36(3):253-55.
- Warnakulasuriya S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 2009; 45:309–16.
- Abbey LM, Kaugars GE, Gunsolley JC, Burns JC, Page DG, Svirsky JA et al. Intraexaminer and interexaminer reliability in the diagnosis of oral epithelial dysplasia. Oral Surg Oral Med Oral Pathol Oral Radiol Endod.1995; 80(2): 188-91.
- Karabulut A, Reibel J, Therkildsen MH, Praetorius F, Nielsen HW, Dabelsteen E. Observer variability in the histologic assessment of oral premalignant lesions. J Oral Pathol Med. 1995; 24(5):198-200.
- Tabor MP, Braakhuis BJ, van der Wal JE, van Diest PJ, Leemans CR, Brakenhoff RH et al. Comparative molecular and histological grading of epithelial dysplasia of the oral cavity and the oropharynx. J Pathol. 2003; 199:354-60.
- Fischer DJ, Epstein JB, Morton TH, Schwartz SM. Interobserver reliability in the histopathologic diagnosis of oral pre-malignant and malignant lesions. J Oral Pathol Med. 2004; 33(2):65-70.
- Abbey LM, Kaugars GE, Gunsolley JC, Burns JC, Page DG, Svirsky JA et al. The effect of clinical information on the histopathologic diagnosis of oral epithelial dysplasia. Oral Surg Oral Med Oral Pathol Oral Radiol Endod.1998; 85(1):74-7.
- Brothwell DJ, Lewis DW, Bradley G, Leong I, Jordan RC, Mock D et al. Observer agreement in the grading of oral epithelial dysplasia. Community Dent Oral Epidemiol. 2003; 31(4):300-5.
- Kujan O, Khattab A, Oliver RJ, Roberts SA, Thakker N, Sloan P. Why oral histopathology suffers inter-observer variability on grading oral epithelial dysplasia: an attempt to understand the sources of variation. Oral Oncol. 2007; 43(3):224-31.
- Amin MB, Greene FL , Edge SB, Compton CC, Gershenwald JE, Brookland RK et al. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more "personalized" approach to cancer staging. CA Cancer J Clin. 2017; 67(2):93-9.
- Forastiere A, Koch W, Trotti A, Sidransky D. Head and neck cancer. N Engl J Med. 2001; 345(26):1890-900.
- Haraf DJ, Rosen FR, Stenson K, Argiris A, Mittal BB, Witt ME et al. Induction chemotherapy followed by concomitant TFHX chemoradiotherapy with reduced dose radiation in advanced head and neck cancer. Clin Cancer Res. 2003; 9(16 Pt 1):5936-43.
- Bernier J, Domenge C, Ozsahin M, Matuszewska K, Lefèbvre JL, Greiner RH et al European Organization for Research and Treatment of Cancer Trial 22931. Postoperative irradiation with or without concomitant chemotherapy for locally advanced head and neck cancer. N Engl J Med. 2004; 350(19): 1945-1952.
- Cooper JS, Pajak TF, Forastiere AA, Jacobs J, Campbell BH, Saxman SB et al. Radiation Therapy Oncology Group 9501/Intergroup. Postoperative concurrent radiotherapy and chemotherapy for high-risk squamous-cell carcinoma of the head and neck. N Engl J Med. 2004; 350(19): 1937-44.
- Khuri FR, Shin DM, Glisson BS, Lippman SM, Hong WK. Treatment of patients with recurrent or metastatic squamous cell carcinoma of the head and neck: current status and future directions. Semin Oncol. 2000; 27(4 Suppl 8): 25-33.
- Campbell PJ, Pleasance ED, Stephens PJ, Dicks E, Rance R, Goodhead I et al. Subclonal phylogenetic structures in cancer revealed by ultra-deep sequencing. Proc Natl Acad Sci U S A. 2008; 105(35): 13081-6.
- Stephenson J. Researchers optimistic about sea change in cancer treatment. JAMA. 2001; 285(22): 2841-2842.
- Dionne KR, Warnakulasuriya S, Zain RB and Cheong SC. Potentially malignant disorders of the oral cavity: current practice and future directions in the clinic and laboratory. Int J Cancer. 2015;136(3):503-15.
- Napier SS, Speight PM. Natural history of potentially malignant oral lesions and conditions: an overview of the literature. J Oral Pathol Med. 2008; 37(1):1-10.
- Warnakulasuriya S, Johnson NW, Van der Waal I. Nomenclature and classification of potentially malignant disorders of the oral mucosa. J Oral Pathol Med. 2007; 36(10):575-80.
- Van der Waal I. Potentially malignant disorders of the oral and oropharyngeal mucosa; terminology, classification and present concepts of management. Oral Oncol. 2009; 45(4-5):317-23.
- Van der Waal I. Oral potentially malignant disorders: Is malignant trans-formation predictable and preventable? Med Oral Patol Oral Cir Bucal. 2014; 19 (4):e386-90.
- Warnakulasuriya S, Kovacevic T, Madden P, Coupland VH, Sperandio M, Odell E et al. Factors predicting malignant transformation in oral potentially malignant disorders among patients accrued over a 10 year period in South East England. J Oral Pathol Med. 2011; 40: 677-83.
- Warnakulasuriya S, Reibel J, Bouquot J, Dabelsteen E. Oral epithelial dysplasia classification systems: predictive value, utility, weaknesses and scope for improvement. J Oral Pathol Med. 2008; 37(3):127-33.
- Jaber MA, Porter SR, Gilthorpe MS, Bedi R, Scully C. Risk factors for oral epithelial dysplasia--the role of smoking and alcohol. Oral Oncol. 1999; 35(2):151-6.
- Odze RD, Maley CC. Neoplasia without dysplasia: lessons from Barrett esophagus and other tubal gut neoplasms. Arch Pathol Lab Med. 2010; 134(6):896-906.
- Greaves M and Maley CC. Clonal evolution in cancer. Nature. 2012; 481(7381):306-313.
- Reid BJ, Li X, Galipeau PC, Vaughan TL. Barrett's oesophagus and oesophageal adenocarcinoma: time for a new synthesis. Nat Rev Cancer. 2010; 10(2): 87-101.
- Jones S, Chen WD, Parmigiani G, Diehl F, Beerenwinkel N, Antal T et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Nat Acad Sci U S A. 2008; 105(11): 4283-88.
- Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010; 467(7319): 1114-7.
- Maley CC, Galipeau PC, Li X, Sanchez CA, Paulson TG, Blount PL, Reid BJ. The combination of genetic instability and clonal expansion predicts progression to esophageal adenocarcinoma. Cancer Res. 2004; 64(20):7629-33.
- Vogelstein B, Kinzler KW. The Multistep Nature of Cancer. Trends Genet. 1993; 9(4):138-141.
- Loeb LA. Mutator Phenotype May Be Required for Multistage Carcinogenesis. Cancer Res. 1991; 51(12): 3075-3079.
- Rajagopalan H, Nowak MA, Vogelstein B, Lengauer C. The significance of unstable chromosomes in colorectal cancer. Nat Rev Cancer. 2003; 3(9): 695-701.
- Sieber OM, Heinimann K, Tomlinson IPM. Genomic instability - the engine of tumorigenesis? Nat Rev Cancer. 2003; 3(9): 701-708.
- Loeb LA, Loeb KR Anderson JP. Multiple mutations and cancer. Proc Natl Acad Sci U S A. 2003; 100(3): 776-81.
- Stratton MR. Exploring the genomes of cancer cells: progress and promise. Science. 2011; 331(6024): 1553-8.
- Gatenby RA, Gillies RJ. A microenvironmental model of carcinogenesis. Nat Rev Cancer. 2008; 8(1): 56-61.
- Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008; 322(5906): 1377-80.
- Notta F, Mullighan CG, Wang JC, Poeppl A, Doulatov S, Phillips LA et al. Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature. 2011; 469(7330): 362-7.
- Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, Colman SM et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011; 469(7330): 356-61.
- Agrawal N, Jiao Y, Bettegowda C, Hutfless SM, Wang Y, David S, et al. Comparative genomic analysis of esophageal adenocarcinoma and squamous cell carcinoma. Cancer Discov. 2012; 2(10): 899-905.
- Dulak AM, Stojanov P, Peng S, Lawrence MS, Fox C, Stewart C et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet. 2013; 45(5): 478-86.
- Farragher SM, Tanney A, Kennedy RD, Paul Harkin D. RNA expression analysis from formalin fixed paraffin embedded tissues. Histochem Cell Biol. 2008; 130(3): 435-445.
- Gnanapragasam VJ. Unlocking the molecular archive: the emerging use of formalin-fixed paraffin-embedded tissue for biomarker research in urological cancer. BJU Int. 2010; 105(2): 274-278.
- Liu A and Xu X. MicroRNA isolation from formalin-fixed, paraffin-embedded tissues. Methods Mol Biol. 2011; 724: 259-67.
- Lu X, van der Straaten T, Tiller M, Li X. Evidence for qualified quantitative mRNA analysis in formalin-fixed and paraffin-embedded colorectal carcinoma cells and tissue. J Clin Lab Anal. 2011; 25(3): 166-73.
- Ludyga N, Grünwald B, Azimzadeh O, Englert S, Höfler H, Tapio S et al. Nucleic acids from long-term preserved FFPE tissues are suitable for downstream analyses. Virchows Archiv. 2012; 460(2): 131-140.
- Stewart GD, Baird J, Rae F, Nanda J, Riddick AC, Harrison DJ. Utilizing mRNA extracted from small, archival formalin-fixed paraffin-embedded prostate samples for translational research: assessment of the effect of increasing sample age and storage temperature. Int Urol Nephrol. 2011; 43(4): 961-967.
- Waldron L, Simpson P, Parmigiani G, Huttenhower C. Report on emerging technologies for translational bioinformatics: a symposium on gene expression profiling for archival tissues. BMC Cancer. 2012; 12:124.
- Campos PF, Gilbert TM. DNA extraction from formalin-fixed material. Methods Mol Biol. 2012; 840:81-5.
- Kalmar A, Wichmann B, Galamb O, Spisak S, Toth K, Leiszter K, et al. Gene-expression analysis of a colorectal cancer-specific discriminatory transcript set on formalin-fixed, paraffin-embedded (FFPE) tissue samples. Diagn Pathol. 2015; 10:126.
- Potluri K, Mahas A, Kent MN, Naik S, Markey M. Genomic DNA extraction methods using formalin-fixed paraffin-embedded tissue. Anal Biochem. 2015;486:17-23.
- Shi SR, Cote RJ, Wu L, Liu C, Datar R, Shi Y, et al. DNA extraction from archival formalin-fixed, paraffin-embedded tissue sections based on the antigen retrieval principle: heating under the influence of pH. J Histochem Cytochem. 2002; 50(8):1005-11.
- Weiss AT, Delcour NM, Meyer A, Klopfleisch R. Efficient and cost-effective extraction of genomic DNA from formalin-fixed and paraffin-embedded tissues. Vet Pathol 2011; 48(4):834-8.
- Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genetics. 2010; 42(9): 790-3.
- Saunders CJ, Miller NA, Soden SE, Dinwiddie DL, Noll A, Alnadi NA et al. Rapid Whole-Genome Sequencing for Genetic Disease Diagnosis in Neonatal Intensive Care Units. Sci Transl Med. 2012; 4(154): 154ra135.
- Trujillano D, Ramos MD, González J, Tornador C, Sotillo F, Escaramis G et al. Next generation diagnostics of cystic fibrosis and CFTR-related disorders by targeted multiplex high-coverage resequencing of CFTR. J Med Genet. 2013; 50(7): 455-62.
- Ziogas DE, Baltogiannis G, Spiliotis J, Tzaphlidou M, Roukos DH. Genome-based diagnostics and predictive tools: a new epoch for breast cancer management. Future Oncol. 2012; 8(10):1211-4.
- Mok SC, Bonome T, Vathipadiekal V, Bell A, Johnson ME, Wong KK et al. A gene signature predictive for outcome in advanced ovarian cancer identifies a survival factor: microfibril-associated glycoprotein 2. Cancer Cell. 2009; 16(6):521-32
- Kerick M, Isau M, Timmermann B, Sültmann H, Herwig R, Krobitsch S et al. Targeted high throughput sequencing in clinical cancer settings: formaldehyde fixed-paraffin embedded (FFPE) tumor tissues, input amount and tumor heterogeneity. BMC Med Genomics. 2011; 4:68.
- Schweiger MR, Kerick M, Timmermann B, Isau M. The power of NGS technologies to delineate the genome organization in cancer: from mutations to structural variations and epigenetic alterations. Cancer Metastasis Rev. 2011; 30(2):199-210.
- Menon R, Deng M, Boehm D, Braun M, Fend F, Boehm D et al. Exome enrichment and SOLiD sequencing of formalin fixed paraffin embedded (FFPE) prostate cancer tissue. Int J Mol Sci. 2012;13(7):8933-42.
- Brakenhoff RH. Cancer. Another NOTCH for cancer. Science. 2011; 333(6046):1102-3.
- Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009; 361(2):123-34.
- Rosenberg NA, Pritchard JK, Weber JL, Cann HM, Kidd KK, Zhivotovsky LA et al. Genetic structure of human populations. Science. 2002; 298(5602):2381-5.
- Ellis MJ, Perou CM. The genomic landscape of breast cancer as a therapeutic roadmap. Cancer Discov. 2013; 3(1):27-34.
